


盛荣:靶向核受体的抗前列腺癌小分子药物研发新进展

盛荣
浙江大学副教授,浙江大学金华研究院小分子创新药团队负责人,浙江省新药审评专家,浙江省杰出青年基金获得者,浙江省“151”人才,《中国现代应用药学》编委,浙江省药学会药化和抗生素专委会委员。盛荣副教授针对肿瘤、病毒、炎性疾病等重大疾病,将计算机辅助药物设计(CADD)、蛋白降解靶向嵌合体(PROTAC)等前沿技术与经典药物设计相融合,开展基于靶点的新药研发和转化研究,发现临床候选分子(PCC),并开展高端仿制药的非专利绿色工艺、新晶型、新盐型研究,协助企业报批生产。近年来以第一、通信作者在 J Med Chem,Brief Bioinform,Org Lett 等期刊共发表 SCI 论文 70 余篇,获得授权美国专利 1 项,中国发明专利 23 项。作为主要研制人员研发的 APG-1387 和 BIOS-0618 分别进入Ⅱ期和Ⅰ期临床研究;主持国家自然科学基金 5 项,国家新药创制重大专项、国家支撑计划、省科技厅领雁项目、省科技厅 重点项目、省自然科学基金重点、省自然杰出青年基金等,研究成果荣获浙江省科技进步二等奖 4 项,三等奖 1 项。
靶向核受体的抗前列腺癌小分子药物研发新进展
袁乐儿 1#,廖金标 1#,蔡吕涛 1, 2 ,胡陈娴 1 ,杨号东 2 ,盛荣 1, 2*
(1浙江大学药学院,浙江 杭州 310058;2. 浙江大学金华研究院,浙江 金华 321002)
[ 摘要 ] 作为全球男性第二大常见癌症,前列腺癌严重威胁着人类健康。在前列腺癌发病过程中,雄激素受体(androgen receptor, AR)起着关键作用,糖皮质激素受体(glucocorticoid receptor,GR)也参与调节部分 AR 通路下游基因,因此,阻断 AR 和 GR 信号通路是前列腺癌治疗的重要策略。综述从核受体 AR 和 GR 的结构特征和生物学功能出发,从药物化学角度系统介绍了近 5 年来靶向核受体的抗前列腺癌药物研发新进展,包括雄激素竞争性 AR 拮抗剂、雄激素非竞争性 AR 拮抗剂、AR 降解剂、GR 拮抗剂和 AR/GR 双重拮抗剂等,为去势抵抗性前列腺癌的治疗提供新思路。
2022 年全球癌症统计资料显示,前列腺癌(prostate cancer,PCa)已成为男性发病率第 2 的癌症,致死率排名第 5[1]。前列腺癌发病机制复杂,影响因素众多。研究表明,核受体家族蛋白在前列腺癌的发生发展中具有重要作用,核受体主要有雄激素受体(androgen receptor,AR)、糖皮质激素受体(glucocorticoid receptor,GR)和盐皮质激素受体(mineralocorticoid receptor,MR)等。其中, AR 信号通路在前列腺癌发生和发展过程中起关键作用 [2],拮抗 AR 通路是治疗激素敏感性前列腺癌及去势抵抗性前列腺癌(castration-resistant prostate cancer,CRPC)的重要策略。目前,第 2 代 AR 拮抗剂恩杂鲁胺已成为一线治疗药物,但随着 AR 拮抗剂的广泛使用,临床耐药已经产生,耐药机制包括 AR 基因扩增、AR 配体结合域上的点突变和缺乏配体结合域(ligand-binding domain,LBD)的剪接变体(AR variants,ARVs)产生 [3]。
临床研究发现,CRPC 的耐药往往伴随着 GR 表达的上调。由于 AR 和 GR 高度相似,在 CRPC 中,GR 可绕过 AR 阻断,代替 AR 调控 AR 蛋白的表达,促进肿瘤细胞增殖 [4]。研究表明,GR 拮抗剂可恢复前列腺癌细胞对 AR 拮抗剂的敏感性,因此,同时阻断 AR 和GR 信号通路成为克服 CRPC 耐药性的有效策略 [5]。
本文对近 5 年(2018—2023)靶向核受体 AR 和 GR 的抗前列腺癌药物研发新进展进行综述,包括雄激素竞争性 AR 拮抗剂、雄激素非竞争性 AR 拮抗剂、AR 降解剂、GR 拮抗剂和 AR/GR 双重拮抗剂,旨在为 CRPC 的治疗提供新思路。
1 雄激素受体
1.1 雄激素受体的结构及功能
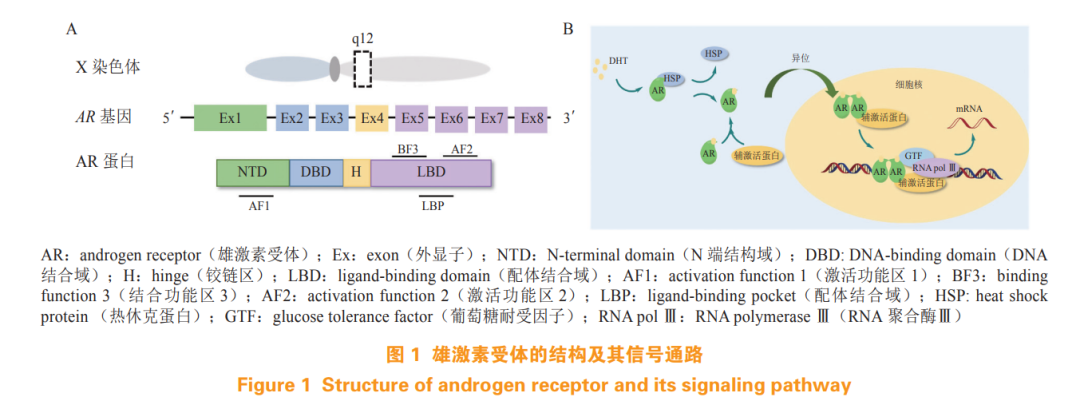
AR 是由 919 个氨基酸组成的类固醇受体转录因子,为核受体家族成员。AR 基因位于染色体 Xq12 上,由 8 个外显子组成,如图 1A 所示。AR 由 N 端结构域(N-terminal domain,NTD)、DNA 结合域 (DNA-binding domain,DBD)、铰链区(hinge,H) 和 LBD 组成。外显子 1 编码的 NTD 包含激活功能区 1(activation function 1,AF1), 支持 AR 的转录活性。外显子 2 和 3 编码的 DBD 包含 2 个锌指结构,第 1 个锌指结构决定 DNA 结合特异性,第 2 个锌指结构与 AR 二聚化及 DNA 受体复合物的稳定性有关。C 端包含由外显子 4 编码的柔性铰链区和外显子 5 ~ 8 编码的高度保守的 LBD,LBD 包含配体结合口袋(ligand-binding pocket,LBP)、激活功能区 2(activation function 2,AF2)和结合功能区 3(binding function 3,BF3)[6]。
1.2 雄激素受体的信号通路
AR 的信号通路如图 1B 所示,未与雄激素 [ 睾酮或二氢睾酮(dihydrotestosterone,DHT)] 结合的 AR 蛋白位于细胞质中,与热休克蛋白(heat shock protein,HSP)形成稳定的复合物。雄激素与 AR 的 LBD 中的 LBP 结合后,诱导 AR 构象发生变化,随后,AR 与 HSP 分离,发生同源二聚化,并易位至细胞核内,二聚化 AR 的 DBD 与 DNA 上的雄激素应答元件(androgen response element,ARE) 结合后,招募一系列转录共调节因子,进而调节近百种 AR 靶基因的表达,包括前列腺特异性抗原(prostate-specific antigen,PSA)、跨膜丝氨酸蛋白酶 2(transmembrane protease serine 2,TMPRSS2)等,该过程的过度激活促进前列腺癌的进展 [7]。
2 雄激素竞争性雄激素受体拮抗剂
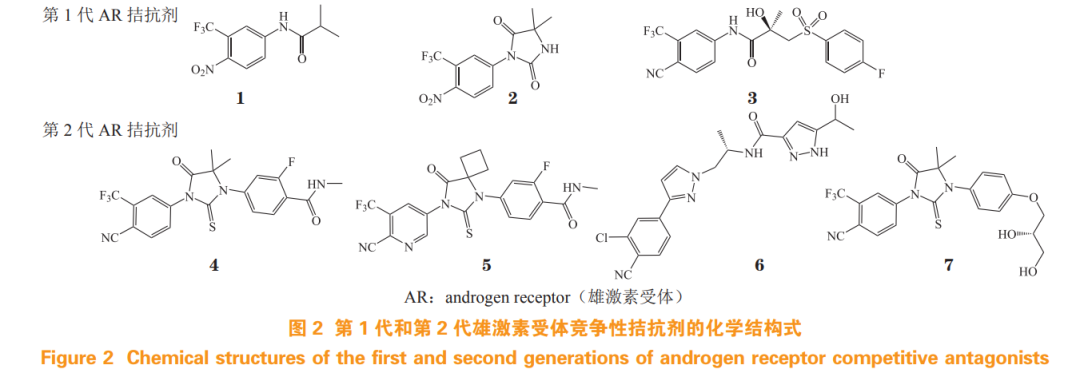
AR 的激活高度依赖于雄激素与 LBP 的结合,因此靶向 AR 的 LBP 拮抗雄激素的作用,是近年 AR 拮抗剂新药研发的主要策略。目前已上市的第 1 代和第 2 代 AR 拮抗剂(结构式见图 2)均结合于 AR LBP 位点,其作用机制为 LBP 与雄激素竞争性结合从而阻断 AR 激活,进而抑制 AR 信号通路, 降低血清 PSA 水平 [6]。
2.1 第 1 代竞争性雄激素受体拮抗剂
氟他胺(flutamide,1)于 1989 年获美国食品药品监督管理局(Food and Drug Administration, FDA)批准用于治疗 PCa,是首个上市非甾体类 AR 拮抗剂。通过进一步的结构优化得到的尼鲁他胺 (nilutamide,2)和比卡鲁胺(bicalutamide,3),分别于 1995 年和 1996 年获批。第 1 代 AR 拮抗剂与 AR 的亲和力较弱,不能充分阻断 AR 通路,与雄激素剥夺疗法(androgen-deprivation therapy, ADT)联用可适度延长患者总生存期,但不良反应发生率较高,且长期用药易引起 AR LBD 的耐药突变,包括 AR W742C/L 和 H875Y/T878A 突变等 [6], 导致拮抗剂转变为部分激动剂,使患者产生耐药性, 最终发展为 CRPC[8]。
2.2 第 2 代竞争性雄激素受体拮抗剂
恩杂鲁胺(enzalutamide,4)是首个第 2 代 AR 拮抗剂,与 AR 结合亲和力显著提升,能抑制 AR 核易位、与雄激素结合以及与靶基因结合的全过程,分别于 2012 年、2018 年和 2019 年被 FDA 批准用于治疗转移性去势抵抗性前列腺癌(metastatic castration-resistant prostate cancer,mCRPC)、非转移性去势抵抗性前列腺癌(non-metastatic castration-resistant prostate cancer,nmCRPC)和激素敏感性前列腺癌(hormone-sensitive prostate cancer,HSPC)[9];但是因在患者脑内恩杂鲁胺稳态水平较高,导致拮抗 γ-氨基丁酸 α(γ-aminobutyric acid α,GABAα)受体从而易产生癫痫等不良反应。阿帕鲁胺 (apalutamide,5)为恩杂鲁胺的类似物,该药对 AR 的拮抗活性更高,分别于 2018 年和 2019 年获批用于治疗 nmCRPC 和 mHSPC[9],且不易入脑,中枢神经系统(central nervous system,CNS)不良反应较少。但 AR LBD 的 F877L 突变使恩杂鲁胺和阿帕鲁胺转变为激动剂,以及ARVs的产生也导致耐药[10]。
达洛鲁胺(darolutamide,6)由拜耳和奥利安公司联合研发,该药能有效抑制恩杂鲁胺和阿帕鲁胺耐药的 AR F877L 突变体,延长高危 nmCRPC 患者的无转移生存期,于 2019 年获 FDA 批准用于治疗 nmCRPC[11]。但是患者发生疲劳、疼痛等副作用的比例较高,导致临床应用受限 [12]。
恒瑞医药研发的瑞维鲁胺(rezvilutamide,7) 也为恩杂鲁胺类似物,在 mCRPC 的Ⅰ/Ⅱ期临床试验中,该药 160 mg · d-1 剂量与 360 mg · d-1 恩杂鲁胺的药物暴露量相当,且耐受性良好 [ 最大耐受剂量 (maximal tolerance dose,MTD)大于 480 mg ·d-1 ];同时,该药还具有血脑屏障透过率低、诱发癫痫风险小和安全性高等优点 [13]。瑞维鲁胺联合 ADT 治疗高瘤负荷 mHSPC,可显著延长患者总生存期, 于 2022 年 6 月获国家药品监督管理局(National Medical Products Administration,NMPA)批准治疗 mHSPC[14]。
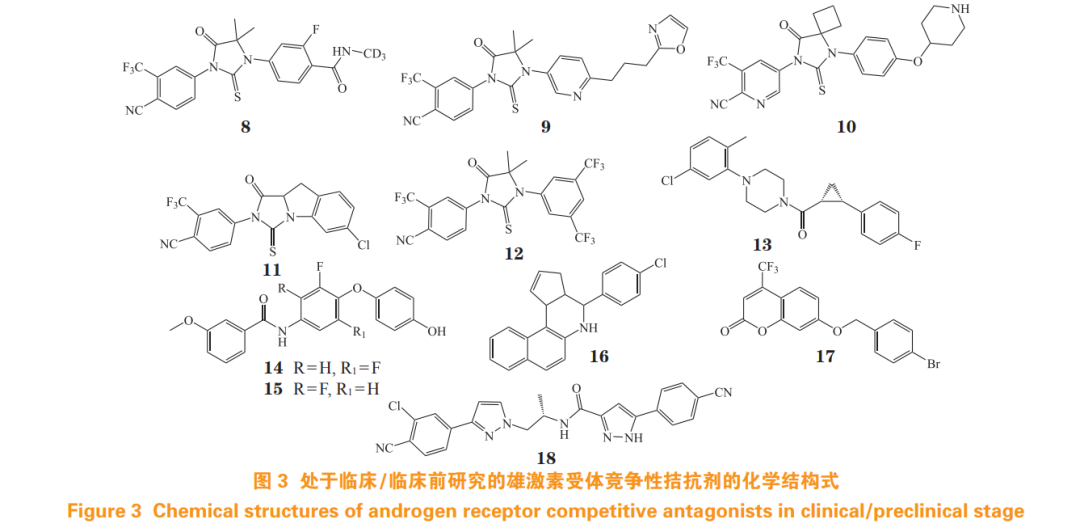
目前,还有多种 AR 拮抗剂处于临床试验阶段(结构式见图 3)。例如,HC-1119(8)为氘代恩杂鲁胺,针对 mCRPC 患者的Ⅰ期临床试验显示, 与恩杂鲁胺相比,HC-1119 代谢缓慢、半衰期更长, 母体药物的血浆浓度增加约 40%,主要代谢物的血浆浓度降低约 75%[15],目前正开展治疗 mCRPC 上市申请。
开拓药业的普克鲁胺(9)与 AR 的结合亲和力比恩杂鲁胺高 3.4 倍 [ 半数抑制浓度(half maximal inhibitory concentration,IC50)为 32 nmol·L-1 ,抑制常数(inhibition constant,Ki)为 14 nmol· L-1 )], 且对 ARF877L,ARW742C/L 和 ARH875Y/T878A 等耐药突变体有效。该药在体内消除速度慢,单次和连续给药的平均表观清除率分别为 0.55~1.30 和 0.17~0.46 L·h -1 [16],且 CNS 分布低,不易诱发癫痫,mCRPC 的Ⅱ期临床试验效果良好 [17]。
西安杨森的 TRC-253(10)可强效抑制野生型 AR 和 F877L 突变体,IC50 分别为 54 和 37 nmol·L-1 ,在F877L突变体的异种移植模型中抑瘤效果显著[18], 正处于 mCRPC Ⅱ期临床 [19]。
目前,正大天晴的 TQB3720 [20]、日本大鹏药品的 TAS-3681 [21]、康朴的 X-Synergy® (结构均尚未披露)处于Ⅰ期临床研究。
此外,还有多种 AR 拮抗剂正处于临床前研究阶段(结构式见图 3)。其中,化合物 11 在 LNCaP 细胞中的 AR 转录抑制活性与恩杂鲁胺相近 [8],化合物 12 在 LNCaP 细胞中的活性较恩杂鲁胺提高近 60 倍 [22]。(+)JJ-450(13)经高通量筛选和结构优化得到 [23],在 C4-2-PSA-rl 细胞中,其活性与恩杂鲁胺相当,作用机制为延缓 AR 核易位,且对恩杂鲁胺耐药的 CRPC 有效,能抑制剪接变体 AR-V7 的转录活性和基因表达 [24]。苯甲酰衍生物 14 和 15,对表达 AR 野生型(wild-type,WT)、T877A 和 H874Y 突变体的前列腺癌细胞,均具有显著的增殖抑制作用 [25]。
侯廷军团队基于 AR LBD 结构,经虚拟筛选发现 AT2(16)为全新骨架 AR 拮抗剂,其抗 AR 转录活性的 IC50 为 0.06 µmol · L-1 ,能抑制 AR 下游靶基因及 AR 核易位 [26]。该团队采用分子动力学模拟构造 AR LBD 二聚体结构,并进行虚拟筛选和结构优化,其中香豆素类衍生物 17 抑制 AR 转录的 IC50 为 0.17 µmol·L-1 ,机制可能是抑制 AR 二聚化 [27]。
以达洛鲁胺为先导化合物,对其 2-氯苯腈部分和吡唑部分进行结构改造,其中,类似物 18 能有效拮抗 AR 转录活性(IC50 = 0.05 µmol· L-1 ), 对 ARF876L 和 ART877A 的效力均优于达洛鲁胺,且抑制 AR 下游靶基因表达 [28]。
3 非雄激素竞争性雄激素受体拮抗剂
AR LBP 易突变的性质严重限制经典 AR 竞争性拮抗剂的临床使用。近年来,靶向 AR 雄激素非竞争性位点的拮抗剂也得到了发展,如靶向 LBD 的 AF2 和 BF3,NTD 的 AF1 以及 DBD 的拮抗剂(结构式见图 4),以寻求克服耐药的前列腺癌新疗法。
3.1 靶向配体结合域激活功能区 2 的拮抗剂
AF2 是 AR 与雄激素结合发生构象变化后在 LBD 表面形成的疏水口袋,在该位点招募共调节因子对 AR 发挥转录活性至关重要。AF2 可通过与共调节因子 LXXLL 和 FXXLF 基序特异性相互作用来募集辅助激活因子。AF2 作为转录激活结构域,在正常细胞中调节功能较弱,而在 CRPC AR 高表达的环境中趋于主导。因此,靶向 AF2 直接阻断 AR 与辅助激活因子相互作用的疗法,理论上不会受 LBP 耐药突变的影响,从而具有良好的临床应用前景[29]。
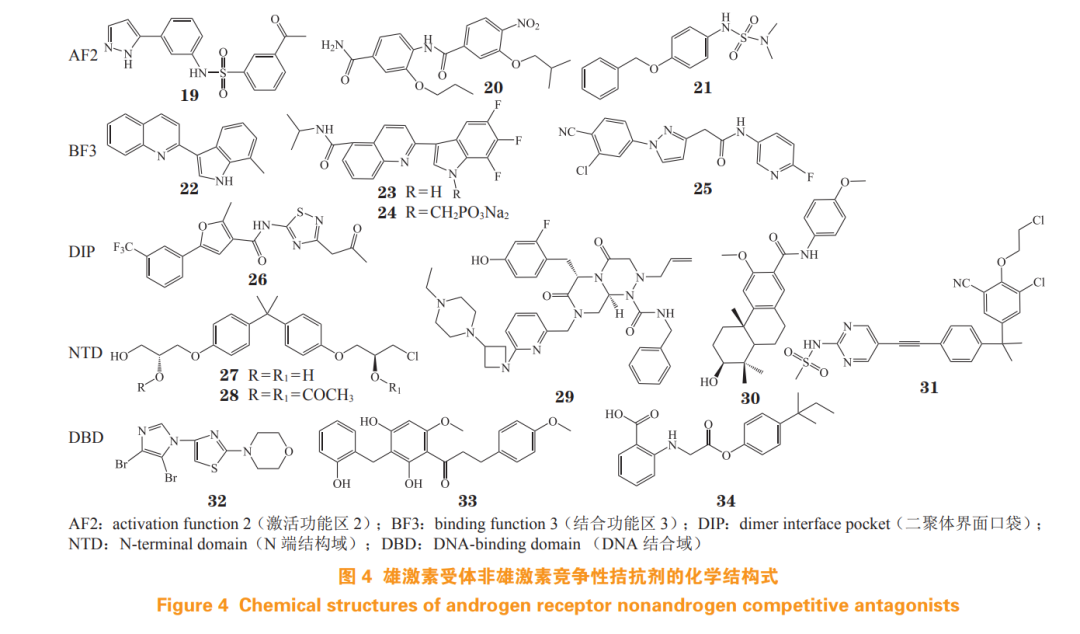
针对 AF2 蛋白的虚拟筛选而发现的苯磺酰胺类化合物 IMB-A6(19),可有效抑制 WT-AR和 AR突变体(ART877A 和 ARF876L)的转录活性。免疫共沉淀实验证明,IMB-A6 靶向 AF2 位点,破坏 AR 与共调节因子脯氨酸-谷氨酸-亮氨酸富集蛋白 1(proline-,glutamic acid- and leucine-rich protein 1, PELP1)在AF2位点的相互作用,从而抑制AR活性[30]。
双苯甲酰胺类衍生物 20 的 AR 转录抑制活性优异,IC50 为 16 nmol· L-1 ,且 100 nmol· L-1 浓度下能有效抑制 LNCaP 细胞活性,并抑制 AR 调控基因 PSA 的表达。机制研究表明,该化合物在 AF2 位点抑制 AR 与共调节因子 PELP1 的相互作用,从而发挥 AR 拮抗活性 [31]。
基于 AF2 蛋白进行虚拟筛选发现的磺酰胺类化合物 T1-12(21),其 AR 转录抑制活性优良,IC50 为 0.47 μmol· L-1 ,能显著抑制 AR 下游基因表达和 AR 核易位,在 LNCaP 异种移植模型的效果与恩杂鲁胺相当。经时间分辨荧光共振能量转移技术(time-resolved fluorescence resonance energy transfer,TR-FRET)AR 共激活因子试剂盒验证,T1-12 能浓度依赖性地下调 FRET 信号,通过阻断 AF2 与共调节因子 FXXLF 结合,从而发挥 AR 拮抗活性 [32]。
3.2 靶向配体结合域结合功能区 3 的拮抗剂
BF3 位点在空间上与 AF2 相邻,BF3 通过招募 FK506 结合蛋白 52(FK506 binding protein 52, FKBP52)和Bcl-2 结合抗凋亡蛋白1(Bcl-2-associated athanogene-1,BAG-1)等辅助激活因子来调控 AR 转录活性。同时,BF3 还与相邻的 AF2 形成变构网络以调节 AR LBD 功能。前列腺癌和雄激素不敏感综合征中大量的 BF3 突变(前者如 Gln670 等;后者如 Leu830,Pro723 等),以及 BF3 抑制剂氟灭酸的发现,证明靶向 BF3 作为治疗前列腺癌策略的可行性 [33]。
Leblanc 等 [34] 发现 2-(1H-吲哚-3-基)喹啉衍生物 VPC-13566(22)具有良好的 AR 转录抑制活性(IC50 = 0.06 μmol·L-1 ),但体内代谢不稳定,半衰期短(T1/2 = 21 min)。对吲哚及喹啉环进行修饰后得到的衍生物 VPC-13789(23),代谢稳定性显著提高(T1/2 = 206 min),能有效抑制WT-AR (IC50=0.19 μmol·L-1 )和 ARF877L 突变体(IC50=0.21 μmol· L-1 )的活性,抑制 LNCaP 细胞的增殖。将 VPC-13789 制备为可口服的磷酸钠前药 VPC-13822 (24),VPC-13822 在 CRPC 异种移植模型中疗效良好,显著降低血清 PSA 水平,且长期毒性小,具有明显的临床价值。
Chen 等 [35] 基于恩杂鲁胺和达洛鲁胺化学结构,经骨架跃迁设计得到一系列衍生物,其中化合物 25 的 AR 转录抑制活性优良(IC50=0.07 μmol·L-1 ),对 ARF877L/T878A 双突变体有效(IC50=0.25 μmol·L-1 ),且能有效抑制 LNCaP 细胞增殖(IC50=6.23 μmol·L-1 ), 在小鼠 LNCaP 异种移植瘤模型中,经口给药(100 mg · kg-1 · d -1 )28 天后,可有效抑制肿瘤生长,分子动力学模拟结果预测该化合物可能结合于BF3位点。
3.3 靶向配体结合域二聚体界面口袋的拮抗剂
侯廷军团队通过分子动力学模拟和小角度 X 射线散射实验,发现 AR 的二聚体界面口袋(dimer interface pocket,DIP),并通过基于结构的虚拟筛选,发现小分子拮抗剂 M17-B15(26)能够有效地破坏 AR 蛋白二聚化,进而抑制 AR 信号转导。其 AR 转录抑制活性优良(IC50=0.03 μmol·L-1 ),且对恩杂鲁胺耐药的 ARF876L/T877A 双突变体有效(IC50=0.15 μmol·L-1 ),能有效抑制 AR 调控及基因转录和翻译水平。在异种移植 LNCaP 细胞模型中,瘤内注射 M17-B15(2.5 mg · kg-1 · d-1 )可显著抑制肿瘤生长。因此,靶向DIP位点是全新的AR拮抗剂研发策略[36]。
3.4 靶向 N 端结构域的拮抗剂
NTD 是 AR 完全转录活性的关键区域,并存在于所有形式的 AR 中,且 NTD 可以在雄激素非依赖性前列腺癌细胞中调节 AR 活性 [21]。因此,靶向 NTD 的拮抗剂有望解决 CRPC 耐药性问题。
从海绵提取物库中筛选而得的 EPI-001 的最有效的立体异构体为 EPI-002(27),该化合物能与 NTD 上的 TAU-5 结合 [37],其乙酰基前药 EPI-506 (28)为首个进入临床的 NTD 拮抗剂,但因药物剂量负担过重、口服生物利用度差而终止试验 [38]。类似物 EPI-7386(29)在 LNCaP 异种移植模型中活性与恩杂鲁胺相当;在恩杂鲁胺耐药的 VCaP 异种移植模型中,EPI-7386 单药或与恩杂鲁胺联用,均表现出显著的抗肿瘤活性,目前处于 mCRPC 的 Ⅰ/Ⅱ期临床试验 [39]。
通过靶向 AR NTD 的虚拟筛选获得的 QW07 (30),具有良好的 AR 转录抑制活性(IC50 = 4.93 μmol·L-1 ),能有效抑制 LNCaP 和 22RV1 细胞增殖。在 CRPC 动物模型中,QW07(10 mg · kg-1 · d-1 )显著抑制 22RV1 和 VCaP 肿瘤的生长。染色质免疫沉淀实验证明,QW07 与 AR NTD 的结合抑制 AR 转录复合物的形成,从而阻止下游基因与启动子、增强子的结合 [40]。
研究发现 DHT 可诱导 AR 入核,并发生液-液相分离,从而形成激活的转录凝集体,该过程主要由 NTD 驱动,采用 ARF877L/T878A 细胞株对化合物库进行筛选,获得化合物 ET0516(31),并经微尺度热电泳等实验证明该化合物结合在 AR NTD。ET0516 可有效地抑制野生型和耐药突变的 AR 的相分离形成,在 5.0 μmol· L-1 浓度下,对 LNCaP 和 VCaP 细胞增殖抑制率大于 50%[41]。
3.5 靶向 DNA 结合域的拮抗剂
DBD 是 AR 与 AREs 结合不可或缺的结构域,对 AR-FL 和雄激素非依赖 ARVs 的核定位至关重要。因此,靶向 DBD 的拮抗剂可以直接阻断 AR 与 DNA 的相互作用,以克服 CRPC 耐药性 [42]。
基于 DBD 结构虚拟筛选发现的噻唑吗啉衍生物 VPC-14449(32)具有优良的 AR 转录抑制活性(IC50 = 0.34 μmol· L-1 ),能有效抑制 LNCaP 细胞和恩杂鲁胺耐药 MR49F 细胞的增殖,生物膜干涉实验结果证明,该化合物选择性地靶向 AR DBD-铰链区结构域表面,进而阻断 AR 与 DNA 相互作用 [43]。
二氢查尔酮衍生物 MF-15(33) 为 AR 和 AKR1C3 的双重抑制剂AKR1C3 为参与雄激素生物合成的酶,与恩杂鲁胺耐药 CRPC 有关。在 10 µmol·L-1 浓度下,MF-15 对 AKR1C3 的抑制率为 87%,并能浓度依赖性地抑制AR-FL和AR-V7的活性,显著抑制 AR 下游 PSA 表达。此外,MF-15 抑制 DBD 中的 P-box 相互作用而发挥 AR 拮抗作用 [44]。
基于 DBD 晶体的虚拟筛选发现了苯甲酸衍生物 Cpd39(34),其 AR 转录抑制活性中等(IC50 = 10.94 μmol·L-1 ),能减少 WT-AR 和 AR-V7 调控基因的表达,且抑制 AR 下游 PSA 基因表达,生物膜干涉实验结果表明,该化合物靶向 DBD-ARE 结合界面位点,抑制 AR DBD-DNA 相互作用 [45]。
4 雄激素受体降解剂
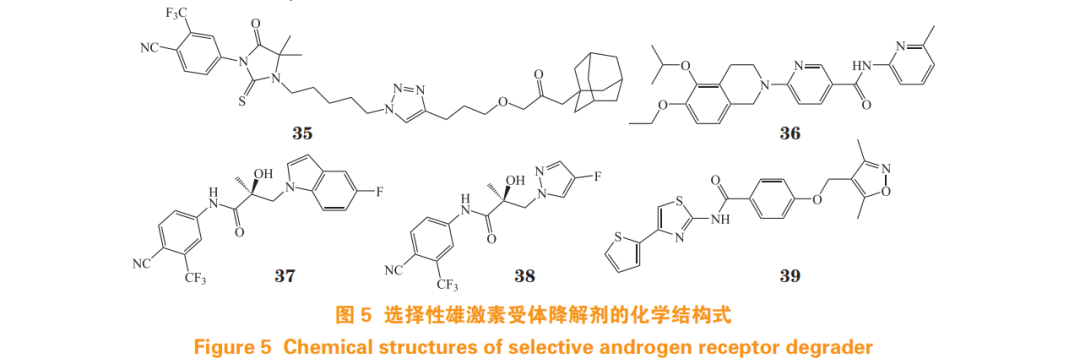
4.1 选择性雄激素受体降解剂
临床患者对 AR 拮抗剂耐药的快速出现,使得对靶向 AR 的新策略需求增加,其中选择性雄激素受体降解剂(selective androgen receptor degrader, SARD)是克服耐药的策略之一(结构式见图 5)[46]。目前,SARD 的确切作用机制尚不明确,一般认为是通过泛素-蛋白酶体系统(ubiquitin-proteasome system, UPS)介导 AR 的降解,可能是通过增强 AR 与 E3 连接酶——双微体同源基因 2(murine double mimute 2,MDM2)的关联而发挥作用。以靶向 LBP 的 AR 拮抗剂 RU59063 母核作为 AR 配体,并通过烷基链和三氮唑连接疏水标签(hydrophobic tag,HyT), 得到化合物 A9(35),其 AR 转录抑制活性良好 (IC50=1.75 μmol·L-1 ), 使用 10 μmol·L-1 A9 处 理 LNCaP 细胞 72 h 后,可基本实现 AR 的完全降解 [47]。
经虚拟筛选发现的二氢异喹啉烟酰胺衍生物 EIQPN(36),其作用位点为 AF1,在 10 μmol·L-1 浓度下,其 AR 抑制率大于 95%,对 LNCaP 细胞增殖抑制率大于 90%。在 NTD 过表达 HEK293T 细胞中,EIQPN 的 IC50 为 0.78 μmol·L-1 。作用机制研究表明,EIQPN 能有效降低 LNCaP,CWR22rv, DU145,PPC1 和 HEK293T 细胞的 AR 和 ARVs 水平, 且能抑制异种移植小鼠模型CWR22rv肿瘤生长[48]。
对 AR 拮抗剂比卡鲁胺采用合环策略,得到类似物 UT-155(37)和 UT-34(38)。作用机制研究表明,两者均能与 AF1 位点结合,并具有 SARDs 功能。体外活性测试表明,在 1.0 μmol·L-1 浓度下,两者在 LNCaP 和 22RV1 细胞系中均能有效降解全长 AR 和 ARVs,并对恩杂鲁胺耐药 MR49F 细胞生长抑制率大于 70%。在 100 mg · kg-1 剂量下,UT-155 和 UT-34 在 LNCaP 去势模型和 MR49F 模型中均能有效抑制肿瘤生长 [49]。
Wu 等 [50] 经药效团虚拟筛选发现了小分子 SARD Z15(39),靶点验证发现该化合物为 AF1 与 LBD 双位点 SARD。体外活性测试表明,Z15 在 LNCaP 细胞中可明显下调 AR 蛋白表达水平,半数最大降解浓度(the half-maximal degradation concentration, DC50)为 1.05 μmol·L-1 。5 μmol·L-1 的 Z15 和环己酰胺(100 μg·mL-1 )联用处理 LNCaP 细胞 24 h 后,可基本实现 AR 完全降解。在 22Rv1 细胞中,Z15 对 AR 和 AR-V7 的 DC50 分别为 1.16 和 2.24 μmol·L-1。
4.2 雄激素受体蛋白降解靶向嵌合体
作为一种新兴的靶蛋白降解技术,蛋白降解靶向嵌合体(proteolysis targeting chimera,PROTAC)近年来迅速发展,引起越来越多的关注。PROTAC 由 3 个部分组成:与 E3 泛素连接酶结合的配体,与靶蛋白(protein of interst,POI)结合的配体以及连接 2 个配体的连接链。PROTAC 分子与 E3 泛素连接酶和 POI 形成三元复合物,特异性诱导 POI 的 泛素化标记,进而通过泛素-蛋白酶体途径降解多泛素化的靶蛋白 [51]。
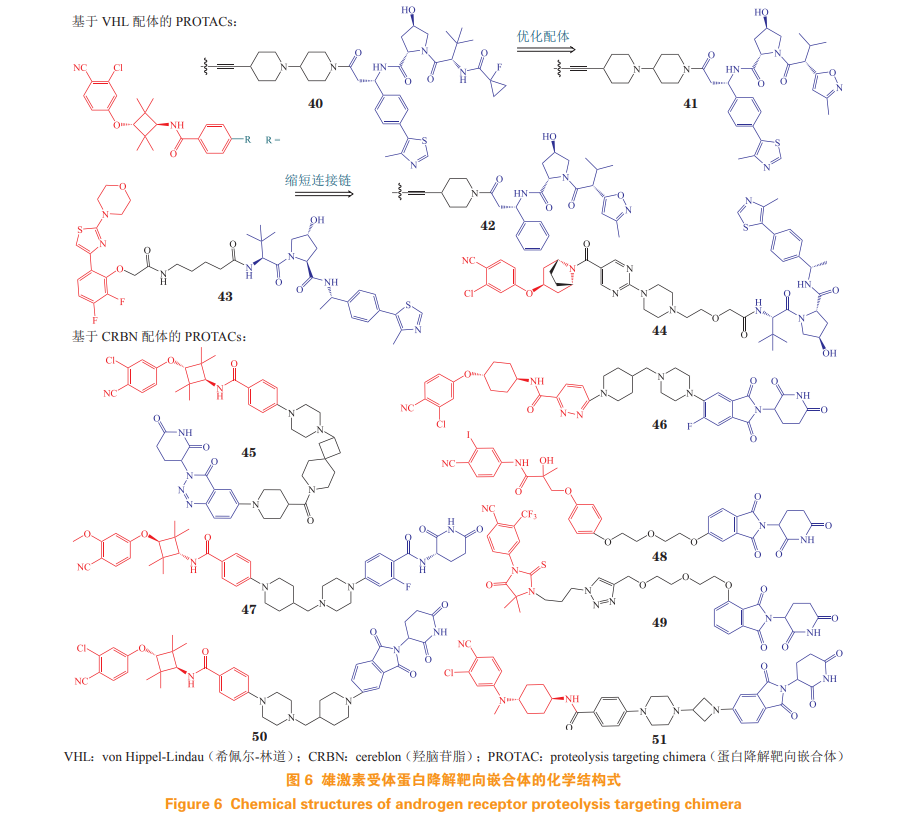
第 1 个靶向 AR 的 PROTAC 分子是含有 E3 泛素连接酶的肽基配体,其理化性质和细胞通透性较差,限制了进一步的应用。2008 年,Crews 等 [52] 首次报道以 MDM2 抑制剂为 E3 连接酶配体,比卡鲁安作为 AR 配体的小分子 AR PROTAC,该化合物能在微摩尔浓度下降解 AR 蛋白,从而开启小分子AR PROTAC 药物(结构式见图 6)时代。
4.2.1 基于希佩尔-林道 E3 泛素连接酶配体的蛋白降解靶向嵌合体 芳氧基环丁基胺衍生物是高活性 AR 拮抗剂 [53],其骨架常作为 AR 配体,以希佩尔-林道(von Hippel-Lindau,VHL)配体为 E3 泛素连接酶配体,通过刚性接头将两者连接得到 PROTAC 分 子 ARD-69(40)[54]。对 ARD-69 的活性进行评估,实验结果表明,ARD-69 在 LNCaP,VCaP 和 22Rv1 细胞中的 DC50 分别为 0.86,0.76 和 10.4 nmol·L-1 。细胞增殖抑制实验中,该化合物对 LNCaP,VCaP 和 22Rv1 细胞的 IC50 分别为 0.25,0.34 和 183 nmol·L-1 , 活性为恩杂鲁胺的 100 倍以上。腹腔给药 50 mg ·kg-1 ARD-69 后,在 48 h 内有效减少 VCaP 异种移植小鼠的 AR 蛋白和 PSA 蛋白。ARD-61(41)是优化 VHL 配体得到的新 PROTAC 分子 [55],表现出良好的体内外 AR 降解活性,且对恩杂鲁胺耐药模型以及 AR 阳性乳腺癌有效。
对 ARD-61 进一步进行结构修饰,将不同 AR 配体与 VHL 配体进行组合得到 ARD-266(42)[56]。ARD-266 在 1 nmol·L-1 浓度下即可诱导 LNCaP, VCaP 和 22Rv1 细胞的 AR 蛋白降解,与 ARD-61 相比,ARD-266 在保持良好的 AR 降解效率和前列腺癌拮抗活性的同时,具有更小的相对分子质量, 且理化性质和成药性更佳。
MTX-23(43)由 DBD 配体、柔性连接链和 VHL 配体组成 [57],由于该化合物结合于 DBD,因 此对 AR-FL 和 AR-V7 具有显著降解活性,DC50 分别为 2.00 和 0.37 µmol·L-1 ,它能有效抑制 CRPC 细胞增殖,经口给药(8.3 mg · kg-1 )显著抑制恩杂鲁胺耐药的前列腺癌异种移植瘤(如 22Rv1 肿瘤),且 MTX-23 对 AR-V7 的降解效果优于 AR-FL,其 深入的机制仍在探索中。
化合物 A031(44)由具有芳氧基托品环的独 特 AR 配体、VHL 配体和包含哌嗪和嘧啶环的连接 体组成。在 VCaP 异种移植斑马鱼中,8.3 μmol·L-1 的化合物 A031 表现出 55% 的肿瘤生长抑制率,与恩杂鲁胺相当,药代动力学性质良好,毒性较低 [58]。
4.2.2 基于羟脑苷脂配体的蛋白降解靶向嵌合体 Takwale 等 [59] 通过使用芳氧基环丁胺 AR 拮抗剂 和羟脑苷脂(cereblon,CRBN)配体得到 TD-802 (45),该化合物是首个报道的具有 CRBN 配体的 AR PROTAC。TD-802 在 LNCaP 细胞中的 DC50 为 12.5 nmol·L-1 ,在体内异种移植物小鼠模型中能有效抑制肿瘤生长,并表现出良好的肝微粒体稳定性和体内药代动力学性质。
ARV-110(46)由阿维纳斯公司研发,经过对 恩杂鲁胺和多种 CRBN 配体及多种连接链的组合 筛选,及进一步结构优化而得,为最早进入临床的 AR PROTAC 分子 [60]。在体内对恩杂鲁胺获得性和内在耐药模型中,经口给予 3.0 mg · kg-1 ARV-110,分别显示出 70% 和 100% 的肿瘤生长抑制率。ARV-110 在Ⅰ期临床试验中用于 mCRPC 患者,具有良好的耐受性、安全性和药代动力学性质[61]。2022年2月, 阿维纳斯公司披露 ARV-110 在治疗 mCRPC 的临床试验中,具有持续抗肿瘤活性和患者获益的证据,在携带 ART878X/H875Y(T878X 为 T878A 或 T878S)突变肿瘤患者中,ARV-110 使 46% 患者的 PSA 水平降低 50% 以上。
目前,ARV-110 正在开展Ⅱ期临床试验 [62]。目前,另有多个 AR PROTAC 已进入临床试 验,其中阿维纳斯公司的 ARV-766(47),相对 于 ARV-110,不仅对 H875Y 和 T878A 等多种突变体亚型具有降解能力,而且能更有效地降解与阿比特龙和其他 AR 途径拮抗剂耐药相关的 L702H 突变体亚型,并在动物模型中得以验证 [63]。2021 年 9 月,ARV-766 在美国开展Ⅱ期临床试验,用于治疗 mCRPC;其结构在 2023 年 4 月的美国癌症研究协会(American Association for Cancer Research,AACR)年会中被披露。此外,百时美施贵宝公司研发的 CC-94676、冰洲石生物科技的 AC-0176 以及海创药业研发的HP518 等 PROTAC 分子都处于Ⅰ期临床试验阶段,用于治疗 mCRPC[64]。
Kim 等 [65] 以比卡鲁胺为 AR 配体,通过柔性 链与 CRBN 配体连接,设计并合成了一系列 AR PROTAC。其中化合物 48 能以剂量和时间依赖性的方式降解 AR 蛋白(LNCaP:DC50=5.21 µmol·L-1 )。另一系列的 AR PROTAC 以恩杂鲁胺衍生物为 AR 配体,通过不同的三氮唑片段连接 CRBN 配体,其中化合物 49 具有优良的 AR 结合亲和力(85%)和AR 降解活性 [66]。
Han 等 [67] 以芳氧基环丁胺为 AR 配体,采用 含哌嗪的连接链与 CRBN 配体沙利度胺相连,分别 得到 ARD-2128(50) 和 ARD-2585(51)。ARD-2128 在 VCaP 和 LNCaP 细胞中的 DC50 分别为 0.28 和 8.3 nmol·L-1 ,能抑制 AR 调控基因,经口给药可有效降低肿瘤组织中 AR 蛋白,有效抑制小鼠肿瘤生长,且毒性较低 [68]。ARD-2585 的降解活性更高,在 VCap 和 LNCaP 细胞中的 DC50 均低于 0.10 nmol·L-1 。ARD-2585 比恩杂鲁胺可更有效地降解全长 AR 和 ARVs,经口生物利用度(小鼠)达 51%,体内疗效 优于恩杂鲁胺,颇具临床应用潜力。
AR PROTAC 为 CRPC 的治疗提供了新的治疗策略,但 AR PROTAC 的相对分子质量较大,成药性亟需提升,实现良好的口服生物利用度具有挑战性。此外,AR PROTAC 多数作用于 LBP 口袋,对于缺乏 LBD 的 ARVs 无效。因此,后续的 AR PROTAC 的研究方向将聚焦解决上述问题。
5 糖皮质激素受体及其拮抗剂的应用
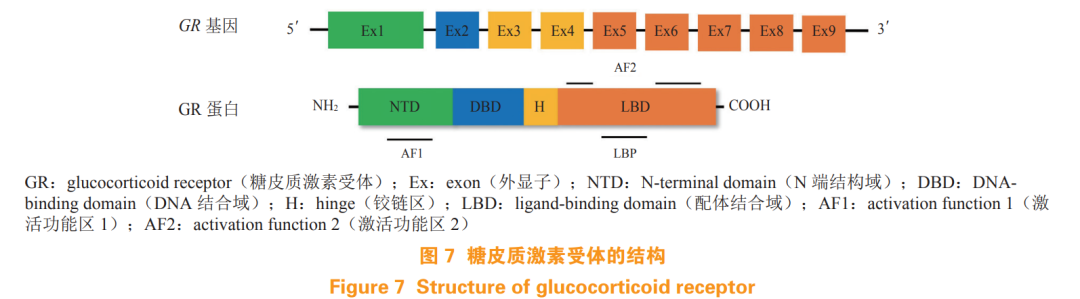
5.1 糖皮质激素受体的结构与功能
人源 GR 基因位于 5 号染色体上,由 9 个外显子组成。如图 7 所示,GR 主要包含 4 个区域, NTD 域由外显子 2 编码,该区域主要负责转录激活功能以及和辅助调节因子结合;DBD 域由外显子 3 和 4 编码,其结构上包含了 2 个锌指结构识别 DNA 上的糖皮质激素反应元件(glucocorticoid-responsive elements,GREs);外显子 5~9 编码铰链区(H) 和 LBD 域,LBD 域包含 1 个配体结合口袋(LBP), 以及 1 个 AF2 结构域,以配体依赖的方式与辅助调节因子相互作用 [69]。
5.2 糖皮质激素受体与去势抵抗性前列腺癌
在前列腺癌中,AR 与 TLE3(transducin-like enhancer of split 3)会和 GR 的增强子相结合;另外,多梳抑制复合物 2(polycomb repressive complex 2,PRC2)会导致组蛋白 3 上的第 27 位赖氨酸的三甲基化(trimethylation of lysine 27 on histone 3, H3K27me3),并通过 zeste 基因增强子同源物 2 (enhancer of zeste homolog 2,EZH2)沉积到 GR 启动子和增强子上,两者共同抑制 GR 的表达 [70]。在早期前列腺癌中,GR 表现为下调趋势,且在 AR 激活的情况下,GR 可能发挥抑制肿瘤发生发展的作用。而在 CRPC 患者中,恩杂鲁胺等 AR 拮抗剂的使用, 使得 AR 的表达受到抑制,并且在治疗过程中 TLE3 会出现表达缺失,进而上调 GR 增强子上的 H3K27 乙酰化,使原先 H3K27 沉积受到抑制,从而恢复 GR 的表达 [70]。而 GR 表达升高后,会与 ARE 结合,共同调控一些经典的 AR 靶基因,导致前列腺癌的生长和恩杂鲁胺耐药的发生。另外,有研究表明 GR 介导的葡萄糖转运蛋白 4(glucose transporter 4,GLUT4)上调与前列腺癌治疗中出现的恩杂鲁胺耐药性和交叉耐药性相关,抑制 GR 或 GLUT4 后,可以减少葡萄糖的摄取,改善癌细胞的耐药性[71]。目前研究认为,GR 和 AR 既存在协同作用,也存在对抗作用,其作用与肿瘤进展关系密切 [72]。综上,GR 具有成为克服 CRPC 耐药治疗关键靶点的潜力,具有器官靶向性的 GR 拮抗剂及 GR/AR 双重拮抗剂(结构式见图 8)可 能在治疗中更具有优势。
5.3 针对糖皮质激素受体过表达的治疗
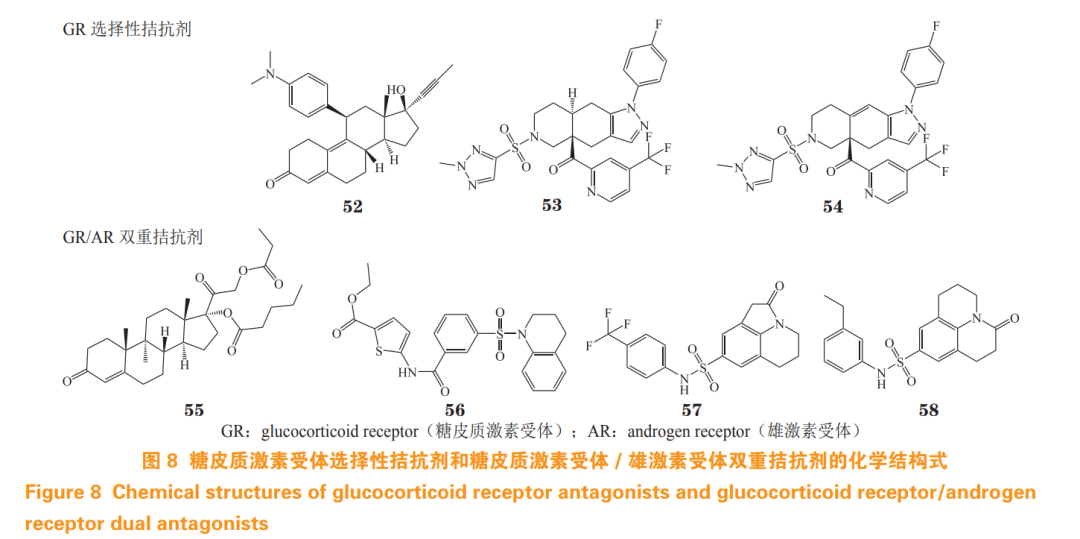
5.3.1 联合糖皮质激素受体拮抗剂用药治疗去势抵抗性前列腺癌 研究发现 GR 拮抗剂可恢复耐药细胞对恩杂鲁胺的敏感性,从而提出联合用药治疗因 GR 过表达引起的 CRPC 耐药。2013 年,米非司酮(mifepristone,52)和恩杂鲁胺联用治疗 CRPC 的临床试验(临床试验编号:NCT 02012296)启动,但因疗效不明显而终止 [72]。2018 年,已开展 GR 选择性拮抗剂 exicorilant(53)和 relacorilant(54)分别与恩杂鲁胺联用治疗 CRPC 的临床试验(临床试验编号:NCT03437941,NCT03674814),但目前临床数据尚未公布。
5.3.2 雄激素受体/糖皮质激素受体双重拮抗剂 联用方式治疗因 GR 过表达而引起的 CRPC 耐药具有一定的意义,但药物-药物相互作用的存在使临床应用受限。因此,靶向 AR/GR 的双重拮抗剂是治疗 GR 过表达 CRPC 的新方向。
对脱氧可的松结构改造所得的化合物 CB-03-10 (55)能有效抑制 AR 和 GR 的转录活性, 对 LNCaP 细胞和恩杂鲁胺耐药 LNCaP-EnzaR 细胞均有效,并能抑制 AR/GR 下游靶基因和蛋白的表达。在小鼠异种移植 LNCaP 细胞模型中,CB-03-10 的活性与紫杉醇相当,具有良好的开发潜力 [73]。
基于 AR LBP 蛋白虚拟筛选发现的化合物 Z19 (56)对 AR 和 GR 均具有良好的转录抑制活性, IC50 分别为 2.03 和 2.50 μmol·L-1 。Z19 能抑制表达 AR 突变 22Rv1 细胞系和 AR 阴性 PC-3 细胞系生长,降低 GR 和 AR 信号下游蛋白和 mRNA 的水平,有效抑制 CRPC 耐药肿瘤的增殖。生物膜干涉实验和分子对接研究表明,该化合物可能靶向 AR 和 GR的 LBP 口袋,与内源性配体竞争性结合 AR/GR 而 发挥拮抗作用 [74]。
基于 AR LBP 蛋白虚拟筛选发现的化合物 H18 (57)具有一定的 AR 拮抗活性,经分子动力学模拟研究及结构优化得到 AR/GR 双重拮抗剂 HD57 (58),该化合物对 AR 和 GR 转录抑制 IC50 分别 为 0.39 和 17.81 μmol·L-1 。此外,该分子对大部分 AR 突变体的抑制活性与达洛鲁胺相当,并能抑制 AR 下游靶基因 PSA 的表达和 AR 核易位 [75]。
6 结语与展望
AR 和 GR 与前列腺癌的发生、发展密切相关, 是抗前列腺癌新药研发的重要靶标。不过,目前已上市的 AR 拮抗剂均靶向 AR 的 LBP 位点,且存在交叉耐药性,限制了临床上的使用。因此,靶向 AR 的非 LBP 位点引起越来越多的关注,包括 AR LBD 上的 AF2 和 BF3 位点,AR 的 DIP 口袋,AR 的 NTD 及 DBD 域等,尤其是后 2 个靶点,有望解决 ARVs 缺乏 LBD 域的问题。另一方面,近年来 AR PROTAC 已成为 AR 靶向治疗的热点,ARV-110 和 ARV-766 已分别进入Ⅱ期临床试验,且疗效明显,前景光明。另外,基于 AR DBD 抑制剂的 PROTAC 52 53 54 55 56 57 58 (如 MTX-23),为治疗 AR-V7 导致的 CRPC 亚群提供了新的解决方案。GR 拮抗剂以及 AR/GR 双重拮抗剂具有独特的作用机制,在 CRPC 治疗领域预期有更广阔的前景。
综上所述,靶向核受体 AR 和 GR 是临床治疗前列腺癌的重要策略,虽然耐药突变、PROTAC 生物利用度低等问题亟需解决,但相信随着计算机辅助药物设计(computer aided drug design,CADD)、人工智能(artificial intelligence,AI)和 PROTAC 技术的进一步发展,将会有更多的靶向核受体抗前列腺癌新药进入临床,为广大患者提供更好的治疗选择。